
July 27, 2019
New software brings lower-resolution cryo-EM maps into focus
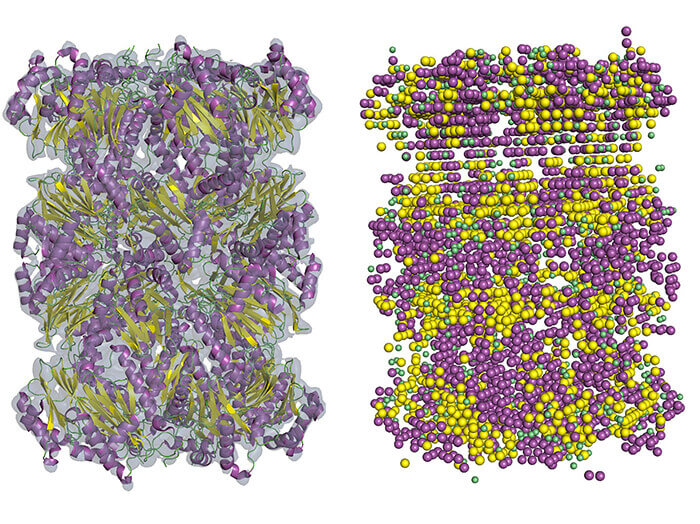 An example of the secondary structure detection in cryo-EM density map using Emap2Sec. A left is an EM map of archaeal 20S proteasome (EMDB ID: EMD-1733). At right are detected secondary structures by Emap2Sec. Points in magenta are the positions of detected alpha helices; yellow points are detected beta strands, and green points are for detected coils (other structures). (Purdue University image/Daisuke Kihara)
Download image
An example of the secondary structure detection in cryo-EM density map using Emap2Sec. A left is an EM map of archaeal 20S proteasome (EMDB ID: EMD-1733). At right are detected secondary structures by Emap2Sec. Points in magenta are the positions of detected alpha helices; yellow points are detected beta strands, and green points are for detected coils (other structures). (Purdue University image/Daisuke Kihara)
Download image
WEST LAFAYETTE, Ind. — Cryo-electron microscopy is now the most popular method for determining protein structures, which helps researchers develop drugs for different kinds of ailments. Over the last several decades, it has replaced X-ray crystallography because it can image proteins that can’t easily be formed into large crystals. The new technique was so revolutionary that it won its developers the 2017 Nobel Prize in chemistry.
The final product of cryo-EM is a map of the density of atoms in biological molecules, but to achieve the level of detail researchers need, they need to conduct further analysis. A new study in the journal Nature Methods outlines a technique to bring low-resolution maps up to par.
The approach researchers use to do this depends on the level of detail they start with. Maps at 2 to 3 ångström (Å, a unit of length used to express the size of atoms and molecules) are generally considered high-resolution. However, maps of this quality are difficult to achieve, and many are still commonly produced in the range of 4 to 10 Å. Of all the proteins deposited to the Electron Microscopy Data Bank from 2016-18, more than 50% were solved at intermediate resolution.
“If the resolution is better than three, then conventional tools can trace amino acid position and build a map of atom positions. But frequently cryo-EM cannot give you a 3 Å map,” said Daisuke Kihara, a professor of biological sciences and computer science at Purdue University. “In maps of 5 Å or lower, you usually can’t see chain connectivity at all.”
Proteins are actually chains of amino acids, and bonding between amino groups and carboxyl groups sometimes creates certain patterns of folding. These patterns, known as alpha helices and beta strands, form the secondary structure of the protein.
In maps from 5 to 8 Å, some fragments of the secondary structure of proteins are usually visible, but tracing the entire chain would be very difficult. Kihara’s new method, known as Emap2sec, uncovers secondary structures in maps from 6 to 10 Å.
Emap2sec has a deep convolutional neural network at the core of its algorithm. These networks are deep-learning systems primarily used to classify images, cluster them by similarity and perform object recognition. It works for protein structure identification in 3D maps because the method “convolves” local map density features to images of a larger region as the information passes through layers of neural network. The local prediction is made in the context of a large region of the map.
Identified secondary structures in 3D maps help researchers to assign known structures of proteins that have already been solved into the map. This means they sometimes have a starting point, or at least a clue of what some of the structure looks like. Emap2sec can help researchers fit their piece into the puzzle more quickly and easily. The identified structure information might also be helpful in finding errors in structure modeling.
The program is available now on GitHub, a software development platform. The research was supported by the National Institutes of Health, National Science Foundation, and Purdue Institute of Drug Discovery.
Writer: Kayla Zacharias
Media contact: Jim Bush, 765-494-2077, jsbush@purdue.edu
Source: Daisuke Kihara, 765-496-2284, dkihara@purdue.edu
Note to Journalists: For a copy of the paper, please contact Jim Bush, Purdue News Service, jsbush@purdue.edu.
ABSTRACT
Protein Secondary Structure Detection in Intermediate Resolution Cryo-EM Maps Using Deep Learning
Sai Raghavendra Maddhuri Venkata Subramaniya, Genki Terashi, and Daisuke Kihara
An increasing number of protein structures have been solved by cryo-electron microscopy (cryo-EM). Although structures determined at near-atomic resolution are now routinely reported, many density maps are still determined at an intermediate resolution, where extracting structure information is still a challenge. We have developed a computational method, Emap2sec, which identifies the secondary structures of proteins (a helices, b sheets, and other structures) in an EM map of 5 to 10 Å resolution. Emap2sec uses a 3D deep convolutional neural network to assign secondary structure to each grid point in an EM map. We tested Emap2sec on 6.0 and 10.0 Å resolution EM maps simulated from 34 structures, as well as on 43 maps determined experimentally at 5.0 to 9.5 Å resolution. Emap2sec was able to clearly identify the secondary structures in many maps tested, and showed substantially better performance than existing methods.
