
December 7, 2017
Research Highlights
Researchers narrow in on cellular causes of enlarged hearts
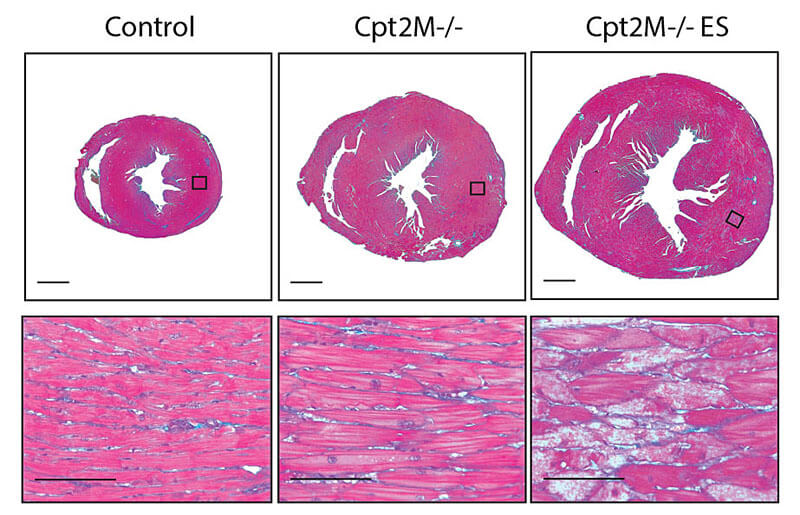 Researchers at Purdue University are working to learn more about enlargement of the heart caused by high blood pressure, diabetes, and obesity. These cross-section images of a mouse heart show the rapid enlargement of a mouse heart (control heart on left, enlarged diseased heart in center, and end-stage diseased heart on right) when it is deficient in an enzyme, carnitine palmitoyltransferase 2 (Cpt2), that is needed for cardiac cells to burn fatty acids as fuel. The research found that using fatty acids as fuel is the key biochemical process for the proper function of the heart. (Purdue University image.)
Download image
Researchers at Purdue University are working to learn more about enlargement of the heart caused by high blood pressure, diabetes, and obesity. These cross-section images of a mouse heart show the rapid enlargement of a mouse heart (control heart on left, enlarged diseased heart in center, and end-stage diseased heart on right) when it is deficient in an enzyme, carnitine palmitoyltransferase 2 (Cpt2), that is needed for cardiac cells to burn fatty acids as fuel. The research found that using fatty acids as fuel is the key biochemical process for the proper function of the heart. (Purdue University image.)
Download image
Study finds that fat fuel is needed to reverse cardiac hypertrophy
WEST LAFAYETTE, Ind. — The metabolic triggers that causes enlarged hearts—a condition known as cardiac hypertrophy—remain elusive, but researchers at Purdue University are getting closer to solving this riddle.
Hypertrophic cardiomyopathy, as the condition is known in medical literature, is often associated with high blood pressure (hypertension), obesity, and Type 2 diabetes (three of the conditions that comprise metabolic syndrome), and can result in heart failure and premature death.
According to Jessica Ellis, assistant professor of nutrition science at Purdue University, an enlarged heart sometimes results from and increased work load, such as in pregnant women or in endurance athletes. More commonly, however, cardiac hypertrophy is caused when the heart cells begin to develop a decreasing ability to use their main source of fuel, fatty acids.
Ellis says in this second, chronic form of cardiac hypertrophy, the metabolism becomes "disrupted, abnormal."
"It's not well understood how this happens," Ellis says, "which is why we don't have any good treatments for hypertrophy."
In the Purdue study, which was published recently in The Journal of Biological Chemistry, the research team used a mouse model in which the mice were unable to produce an enzyme used by cardiac cells to burn fatty acids as fuel.
"We believe we found that this fatty acid biochemical pathway is the key linchpin in this process," Ellis says. "What we think this is showing is that we have to fix the cells' ability to use fatty acid in order to fix the heart."
In addition to fatty acids, heart tissue can also use ketones bodies, which, like fatty acids is a type of cellular fuel produced by the liver.
Some of the mice in the study were put on a diet that forced their bodies to produce ketones (i.e., a ketogenic diet).
"We thought that if we could improve their metabolism with the ketogenic diet, perhaps it would also improve the cardiac hypertrophy," Ellis says. "But it did not; thus, ketone bodies cannot substitute for fatty acids as fuel to stop cardiac cell growth.
"So we're learning that the proper breakdown of fatty acids is required to maintain heart function during metabolic syndrome, and when this is disrupted, some sort of metabolites accumulate and signal the unwanted cell growth in the heart," she says. "We, and other research groups, are going to be taking a very close look at that pathway and really picking it apart to find out exactly where that happens and what those metabolites are, so we can begin work on treatments."
Writer: Steve Tally, 765-494-9809, steve@purdue.edu, @sciencewriter
Source: Jessica Ellis, jmellis@purdue.edu
ABSTRACT
Loss of cardiac carnitine palmitoyltranserase 2 results in rapamycin-resistant, acetylation-independent hypertrophy
Andrea S. Pereyra, Like Y. Hasek, Kate L. Harris, Alycia G. Berman, Frederick W. Damen, Craig J. Georgen, Jessica M. Ellis
Purdue University, West Lafayette, Indiana, USA
Cardiac hypertrophy is closely linked to impaired fatty acid oxidation, but the molecular basis of this link is unclear. Here, we investigated the loss of an obligate enzyme in mitochondrial long-chain fatty acid oxidation, carnitine palmitoyltransferase 2 (CPT2), on muscle and heart structure, function, and molecular signatures in a muscle- and heart-specific CPT2-deficient mouse (Cpt2M-/-) model. CPT2 loss in heart and muscle reduced complete oxidation of long-chain fatty acids by 87 percent and 69 percent, respectively, without altering body weight, energy expenditure, respiratory quotient, or adiposity. Cpt2M-/- mice developed cardiac hypertrophy and systolic dysfunction, evidenced by a 5-fold greater heart mass, 60–90 percent reduction in blood ejection fraction relative to control mice, and eventual lethality in the absence of cardiac fibrosis. The hypertrophy-inducing mammalian target of rapamycin complex 1 (mTORC1) pathway was activated in Cpt2M-/- hearts; however, daily rapamycin exposure failed to attenuate hypertrophy in Cpt2M-/- mice. Lysine acetylation was reduced by ~50% in Cpt2M-/- hearts, but trichostatin A (TSA), a histone deacetylase inhibitor that improves cardiac remodeling, failed to attenuate Cpt2M-/- hypertrophy. Strikingly, a ketogenic diet increased lysine acetylation in Cpt2M-/- hearts 2.3-fold compared with littermate control mice fed ketogenic diet, yet it did not improve cardiac hypertrophy. Together, these results suggest that a shift away from mitochondrial fatty acid oxidation initiates deleterious hypertrophic cardiac remodeling independent of fibrosis. The data also indicate that CPT2-deficient hearts are impervious to hypertrophy attenuators; that mitochondrial metabolism regulates cardiac acetylation; and that signals derived from alterations in mitochondrial metabolism are the key mediators of cardiac hypertrophic growth.
Researchers determine how alphavirus changes into infectious state
WEST LAFAYETTE, Ind. — A key step in the infectious method of a family of disease-causing viruses has been identified by an international team led by scientists from Purdue University.
The research is described in a recent paper in the Proceedings of the National Academy of Science.
The team studied the structure of the Chikungunya virus using cyro-electron microscopy and was able to determine how it changes from a non-infectious immature state to a mature state that is able to infect cells.
Chikungunya is a mosquito-borne disease that has infected more than 40 million people since it was first discovered in 1953.
Michael Rossmann, Purdue's Hanley Distinuished Professor of Biological Sciences, says finding how the virus transforms is a step toward finding a treatment.
"This is specifically about the Chikungunya virus, but it's probably similar to a lot of other alphaviruses, which are a common disease pathogens. This particular virus has become a major problem in Asia and Africa."
The research found that the virus fuses with the membrane of a host cell, cleaving a specific protein, uncovering a spike, which is hydrophobic and, therefore, very slippery. This spike is inserted into the cell and the virus releases its genetic material.
Rossmann's laboratory first determined the structure of the Chikungunya virus in 2013.
Writers: Kayla Zacharias, kzachar@purdue.edu
Steve Tally, 765-494-9809, steve@purdue.edu
Source: Michael Rossmann, 765-494-4911, mr@purdue.edu
ABSTRACT
Structural studies of Chikungunya virus maturation
Moh Lan Yapa,b, Thomas Klosea, Akane Urakamic, S. Saif Hasana, Wataru Akahatac, and Michael G. Rossmann
Cleavage of the alphavirus precursor glycoprotein p62 into the E2 and E3 glycoproteins before assembly with the nucleocapsid is the key to producing fusion-competent mature spikes on alphaviruses. Here we present a cryo-EM, 6.8-Å resolution structure of an “immature” Chikungunya virus in which the cleavage site has been mutated to inhibit proteolysis. The spikes in the immature virus have a larger radius and are less compact than in the mature virus. Furthermore, domains B on the E2 glycoproteins have less freedom of movement in the immature virus, keeping the fusion loops protected under domain B. In addition, the nucleocapsid of the immature virus is more compact than in the mature virus, protecting a conserved ribosome-binding site in the capsid protein from exposure. These differences suggest that the posttranslational processing of the spikes and nucleocapsid is necessary to produce infectious virus.
